
普通PCR、原位PCR、反向PCR和反转录PCR的 基本原理和操作步骤---反向PCR
1 概述
通常测定一个与已知序列相邻的DNA序列是必要的,例如位于编码DNA的上游和下游两侧的区域,转位因子的插入位点以及克隆于Lambda、科期粒或酵母人工染色体载体上的DNA片段末段的未知序列的探针等。这种末端特异探针在Southern Blot或染色体步移(Chromosome walking)所需的噬菌斑杂交或克隆杂交中都十分有用。为了得到边侧序列的探针,通常采用扩展的PCR方法,使相邻边侧区域得以扩增。
2 PCR与反向PCR的区别
PCR只能扩增两端序列已知的基因片段。如图1所示:

图1 PCR引物扩增方向
反向PCR可扩增中间一段已知序列,而两端序列未知的基因片段。如图2所示:

图2 反向PCR引物扩增方向
3 反向PCR基本原理
反向PCR(Reverse PCR)是常规聚合酶链反应技术的修改和发展,是克隆T-DNA插入位点侧翼DNA序列非常有效的方法。反向PCR可用于研究与已知DNA区段相连接的未知染色体序列,因此又可称为染色体缓移或染色体步移。选择已知序列内部没有切点的限制性内切酶对该段DNA进行酶切,然后用连接酶使带有粘性末端的靶序列环化连接,再用一对反向的引物进行PCR,其扩增产物将含有两引物外未知序列,从而对未知序进行分析研究。
扩增前先用限制性内切酶酶切样品DNA,然后用DNA连接酶连接成一个环状DNA分子。如图3所示:
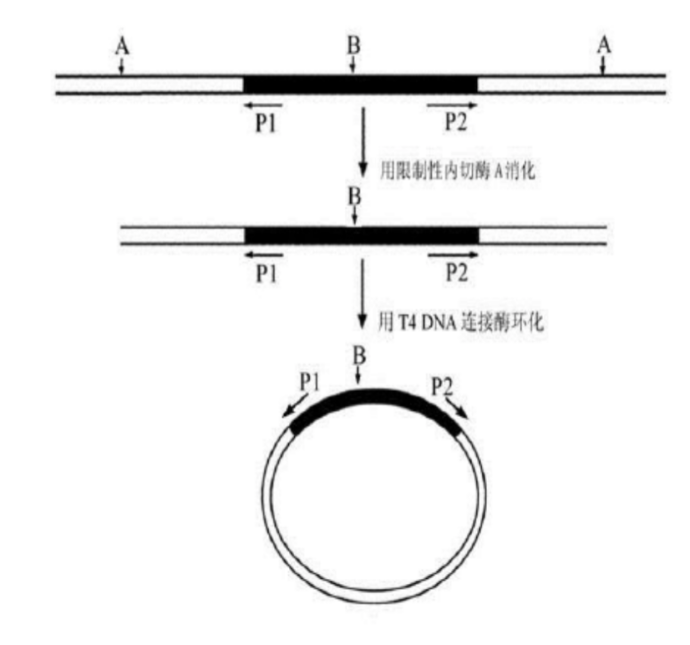
图3 线性DNA的环化
通过反向PCR扩增引物的上游片段和下游片段。如图4所示:
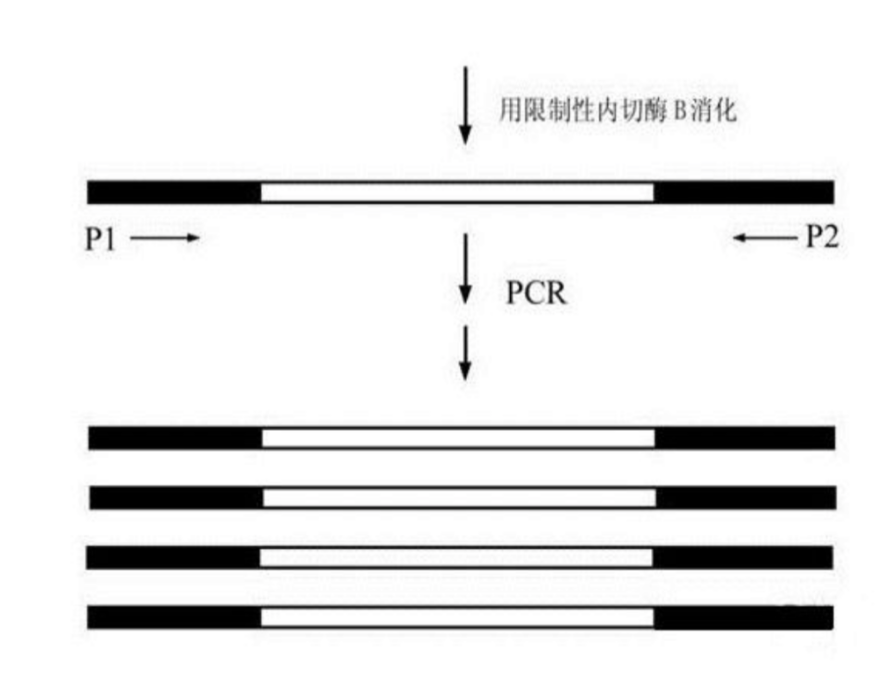
图4 反向PCR扩增
4 反向PCR实验步骤
(1)限制性内切酶消化
① 选择合适的限制性内切酶,基因组一定要切成弥散性的条带。在酶切之前,应对基因组DNA定量。
② 根据T-DNA的左边界序列选择合适的酶切位点(如EcoR I、BamH I、Hind III和Pst I),并在酶切位点上游附近设计引物Z1,Z2,Z3和Z4,然后用这些内切酶分别消化基因组DNA,酶切体系如下:
|
名称 |
体积/ μL |
|
限制性内切酶 |
0.5 (10 u/μL) |
|
限制性内切酶Buffer |
2 |
|
基因组DNA |
3 |
|
ddH2O |
14.5 |
|
Total |
20 |
37℃ 过夜,电泳检测酶切效果。
(2)回收 DNA
① 将酶切产物转到1.5 mL离心管中,用ddH2O洗涤干净,并稀释到250 μL;
② 加入等体积的酚和氯仿(V:V=1:1),涡旋混匀,室温静置1 min;
③ 12,000 rpm离心1 min;
④ 将上清移到另一管中,加入等体积的氯仿,涡旋混匀,室温静置1 min;
⑤ 12,000 rpm离心2 min;
⑥ 将上清移到另一管中,加入2.5倍体积的-20℃预冷的无水乙醇,0.1倍体积的3 M 乙酸钠(pH=5.2),轻轻振荡混匀,室温静置5 min;
⑦ 4℃ 12,000 rpm离心10~15 min;
⑧ 弃上清,尽量除去管壁上的液体;
⑨ 加入1 mL -20℃预冷的70%乙醇,轻轻颠倒几次。12,000 rpm离心5 min;
⑩ 除去乙醇,在超净台上平放,将管壁上的乙醇挥发掉,20~40 μL ddH2O溶解。-20℃保存。
(3) 连接
① 连接体系如下:
|
名称 |
体积/uL |
|
DNA |
2 |
|
10 x T4 DNA Ligase Buffer |
10 |
|
T4 DNA Ligase |
1 |
|
ddH2O |
87 |
|
Total |
100 |
12-14℃,过夜连接。
注:连接体系比较大,而DNA含量比较低,不需加入PEG4000,这样可以促进分子内的连接,减少分子间的连接。
② 连接完成后,65℃水浴10 min灭活。按上述(2)抽提回收,溶解于40 μL ddH2O中。然后就可以用回收的链接片段做PCR了。
5 反向PCR技术的不足
(1)需要从许多酶中选择限制酶,或者说必须选择一种合适的酶进行酶切才能得到合理大小的DNA片段。这种选择不能在非酶切位点切断靶DNA。
(2)大多数有核基因组含有大量中度和高度重复序列,而在YAC或Cosmid中的未知功能序列中有时也会有这些序列,这样,通过反向PCR得到的探针就有可能与多个基因序列杂交。
6反向PCR技术的应用与前景
(1)反向PCR技术适用于基因游走、转位因子和已知序列DNA旁侧病毒整合位点分析等研究,可用于克隆启动子。
(2)反向PCR技术有着自己独特的特点,虽然有不足之处,但它在各学科和各领域都有着重要的作用。随着PCR技术和分子生物学的发展,它在各学科和各领域的作用将会越来越重要,应用也将会更加普遍。




